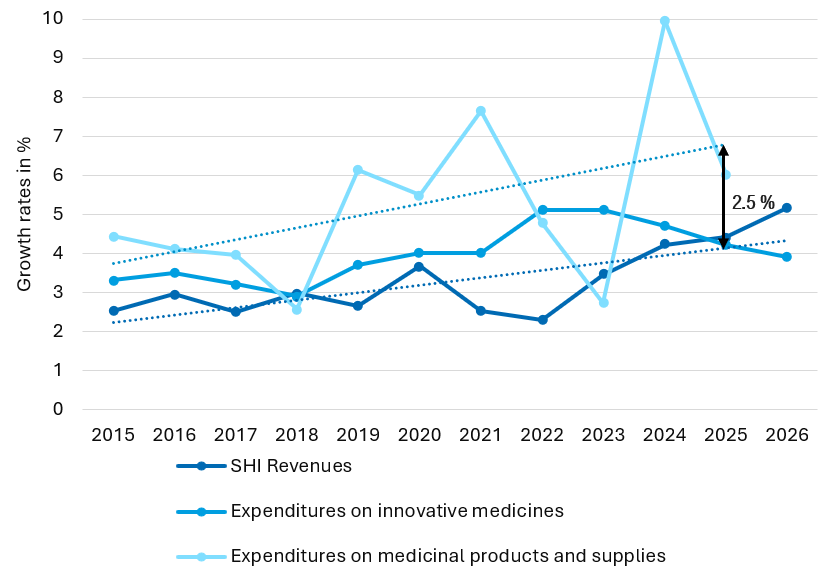
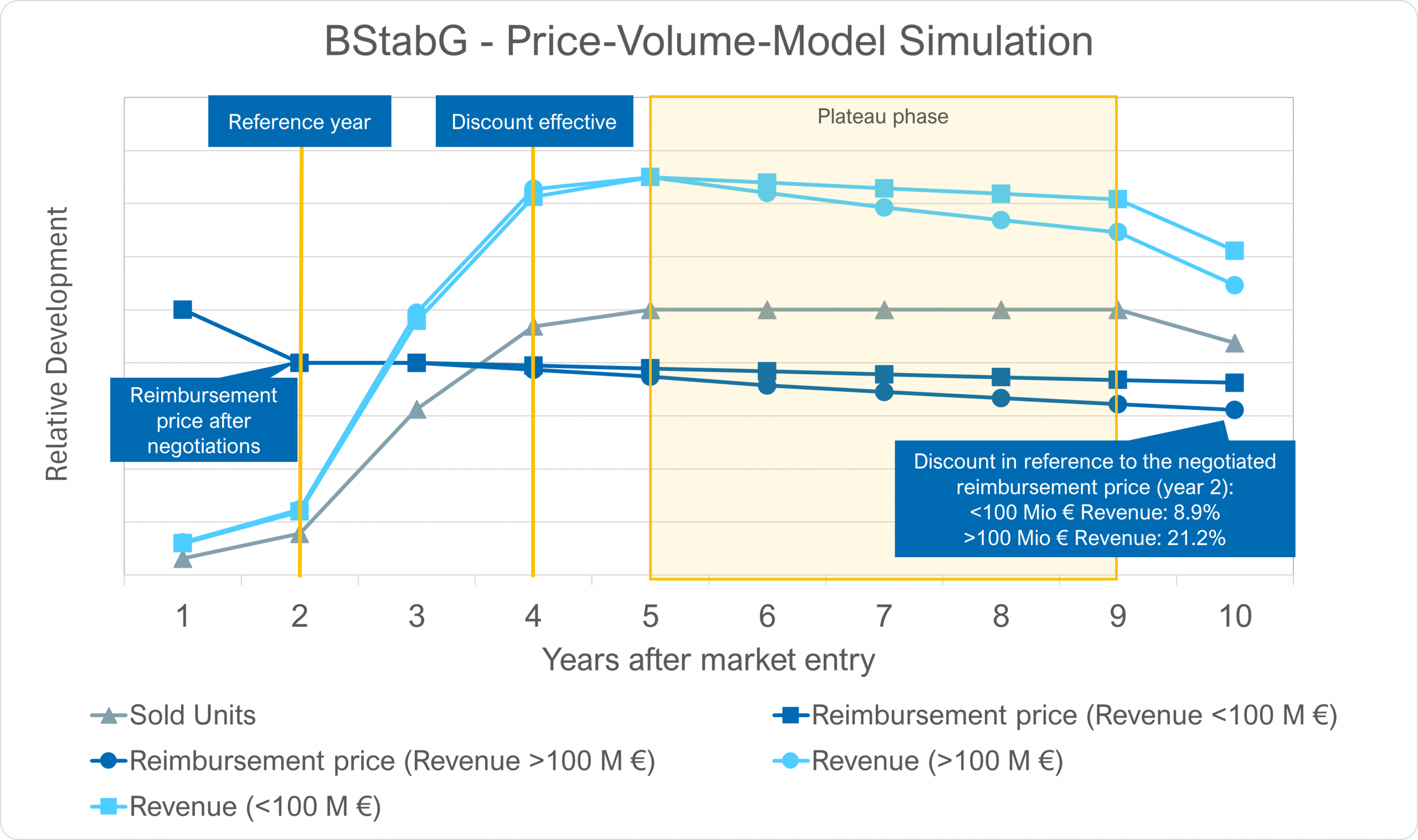
Regulation (EU) 2021/2282
Article 7 (1) of Regulation (EU) 2021/2282 specifies when a health technology must undergo a Joint Clinical Assessment (JCA). Paragraph 1a stipulates that the time of initial market authorization application (MAA) is decisive. A JCA of the new health technology only needs to be carried out if the initial MAA is submitted after the relevant dates specified in Article 7 (2) (oncologics and ATMPs: 12 January 2025, orphan drugs: 13 January 2028, all others: 13 January 2030). Article 7 paragraph 1b further specifies that new indications of existing marketing authorizations will only be subject to a JCA if the health technology has already previously been assessed within a JCA. New indications of health technologies that were initially assessed at national level remain at national level. Therefore, it is to be expected that both procedures will exist in parallel (Figure 1).

Figure 1: National and EU-HTA procedures will exist in parallel even after the relevant dates.
Consequences for pharmaceutical companies
In future, new indications of different health technologies could be assessed at the same time within different procedures (national vs. EU). Thus, it will be important for the pharmaceutical company to ensure the consistency of the national and European assessments. Particularly concerning products with planned submission for marketing authorization in the period of the respective relevant dates, it should be considered whether an early submission before the respective relevant date (consequence: national assessment of all indication extensions) or a later submission (consequence: JCA of all further indication) is strategically more reasonable.
Consequences for the assessment of the added benefit
According to Regulation (EU) 2021/2282, the JCA report should not contain a final assessment of the added benefit, as this is the responsibility of the member states. However, it cannot be ruled out that the decision of the member states may be positively or negatively influenced by the additional information provided in the EU HTA. The consequence would be a positive or negative distortion of the results on the added benefit compared to purely national procedures. To avoid advantages or disadvantages of products of the European procedure compared to products of the national procedure, it must be ensured that both procedures lead to the same result in the case of similar evidence. The national HTA authorities are primarily responsible for ensuring this.